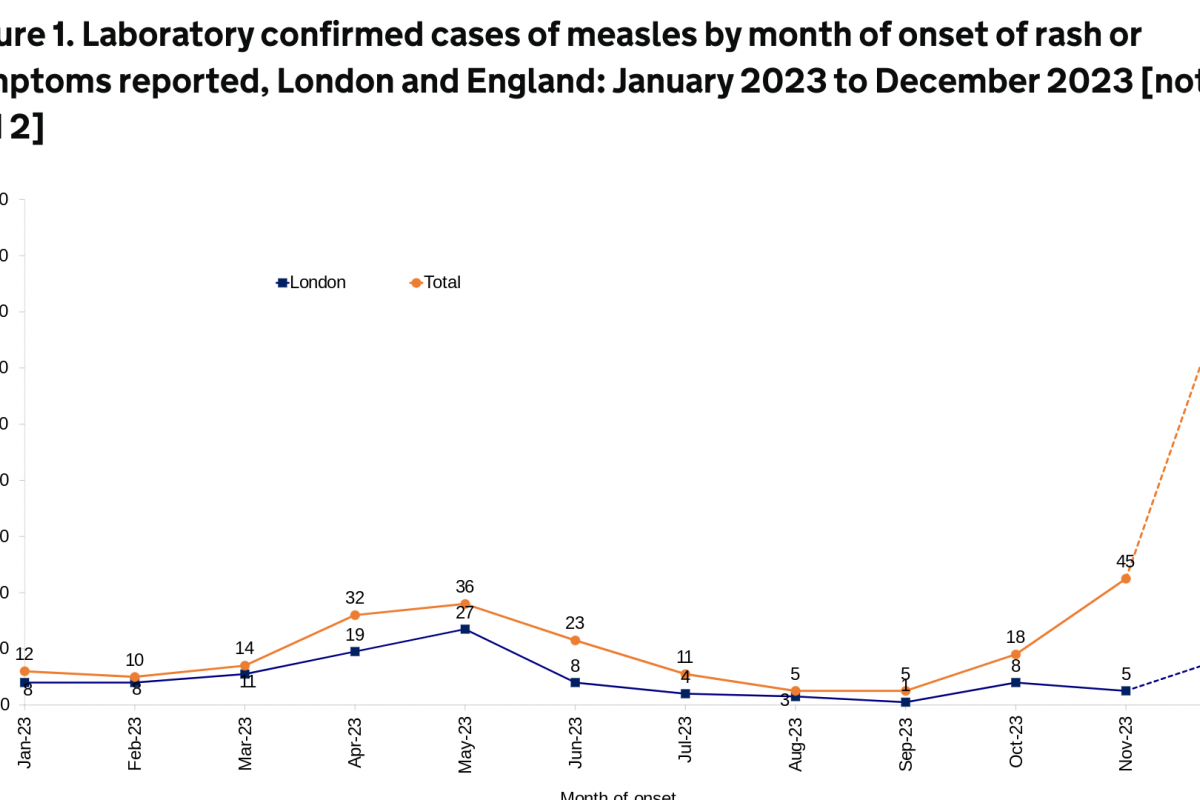
While measles outbreaks have been reported in dozens of countries in 2024, recent attention has focused on the cities of Chicago and London.
The U.K. Health Security Agency (UKHSA) published an updated epidemiological overview on April 18, 2024, stating that an additional 86 cases of measles were confirmed in England last week.
This data brings the total number of confirmed cases since October 2023 to 1,109.
About 39% of the U.K.'s measles cases (76 of 191) were in London during the last four weeks.
In a press release, Dr. Vanessa Saliba, UKHSA Consultant Epidemiologist, commented, "We know some communities in London have very low measles-mumps-rubella (MMR) vaccination rates. The MMR jab offers the best protection against measles."
The effective Priorx MMR vaccine is generally available at clinics and pharmacies in England. However, no measles-only vaccines are offered in England.
In the U.S., the Centers for Disease Control and Prevention (CDC) reported 121 measles cases in eighteen jurisdictions in 2024.
Most of these cases (61) have been reported by the Chicago Department of Public Health over the past two months.
The U.S. CDC republished a global Watch-Level 1, Practice Usual Precautions, Travel Health Notice in March 2024, alerting international travelers of potential health risks and identifying measles outbreaks in 49 countries.
The CDC recommends speaking with a travel vaccine consultant one month before traveling abroad to any outbreak countries.